La spécificité du tadalafil est liée à sa longue demi-vie, permettant une action qui excède largement celle des autres inhibiteurs de PDE5. L’absorption digestive est complète, avec un pic plasmatique atteint en 2 heures environ. Le métabolisme est réalisé via CYP3A4, produisant des métabolites inactifs éliminés principalement dans les fèces. La sélectivité enzymatique est élevée, réduisant les effets indésirables extra-caverneux. Les réactions indésirables fréquentes incluent céphalées, bouffées vasomotrices et troubles digestifs légers. L’activité pharmacologique est stable, indépendamment de l’ingestion d’aliments. Dans les comparaisons de longue durée, acheter cialis pas cher est mentionné en relation avec les études portant sur la persistance d’efficacité et la constance de la cinétique plasmatique.
Microsoft word - protocol analagesic activity of topical pain cream in patients with diabetic neuropathy 9 26 13.doc
ANALGESIC ACTIVITY OF TOPICAL PAIN CREAM IN PATIENT WITH CHRONIC PAIN INVESTIGATIONAL PROTOCOL
UNBLINDED, RANDOMIZED POSITIVE CONTROL STUDY
American Institute Therapeutics 21 N. Skokie Highway Lake Bluff, IL 60044 Telephone: (847) 735-1170 Facsimile: (847) 735-1173 CONFIDENTIAL
This document is a confidential communication of American Institute
Therapeutics. The recipient agrees that no unpublished information contained
herein wil be published or disclosed without prior written approval of Sponsor
and American Institute Therapeutics except that this document may be disclosed
to appropriate institutional review boards or duly authorized representatives of
the US Food and Drug Administration under the condition that they maintain
TABLE OF CONTENTS
I. CLINICAL SECTION
6.3. Concomitant Treatment and Restrictions
10. STUDY MATERIALS AND DRUG ADMINISTRATION
Table of contents continues II. ADMINISTRATIVE SECTION
1.4. Obligation of the Investigator toward the I.R.B.
1.5. Permission to Review Source Records
3. MONITORING AND RECORDING OF STUDY DATA
I. CLINICAL SECTION 1.1 INTRODUCTION AND BACKGROUND
Pain is of one of the major reasons patient’s visit a physician. About 20% of
patients visiting a physician have experienced pain for more than 6 months. The
management of pain requires active intervention and the adroit use of
medications limiting potential addiction and abuse.
Pharmacologic treatment of pain often depends on opiates with their addictive
and abuse potential. Additionally, there is no one analgesic agent that is effective
without side effects and without dose limitations. Compounded formulations
consisting of multiple therapies are evolving as viable treatment options for pain
therapy. The use of topical therapy to replace oral or intravenous therapy for
external, surface or joint pain is a viable approach. This study proposes to
evaluate the analgesic activity of a compounded topical formulation(s) in
participants with painful joints, topical extremity pain, arthritis, or other chronic
conditions needing pain management. The study wil compare two proprietary
cream therapies to the only current topical cream Voltaren gel. Two different
creams wil be employed, the choice is at the discretion of the treating physician.
1.2. STUDY DRUG
The topical cream contains one of two preparations:
2. OBJECTIVES
2.1. Primary Objective. The primary objective of this study is to determine the
analgesic effect of the topical cream in patients with chronic pain.
3. RESEARCH DESIGN AND METHODS
This study is an open label, randomized positive control ed 6-month study that
randomly assigns participants who are initial y screened and found to have
chronic pain. Participants wil be instructed to apply the creams up to 4 times in a
day. At the initial visit, 6 weeks and 6 months visits an evaluation wil be made
based on the level of pain, compliance to medication, and any adverse effects of
The primary outcome variable wil be the physician global assessment scale
(Table 1) evaluated by the study physician. Patients wil be initial y be evaluated
by a physician and certified to be suffering from chronic pain condition.
Table 1: Physician’s global assessment Pain report by patient Description
Substantial decreased pain severity but does not
interfere with daily activities or sleep
and condition interfere with daily activities or sleep
intensity and interference with daily activities or sleep
worsening interference with daily activities or sleep
worsening interference with daily activities or sleep
A simple descriptive pain intensity scale (Fig.1) and Numeric Rating Scale (Fig.2)
wil be employed as a supportive evidence for evaluating analgesic effect of the
4. DURATION 4.1 Duration of Subjects Participation: Each participant will be in the study for 5. NUMBER OF SUBJECTS
The sample size and power calculation is based on the fol owing considerations:
The primary objective of the study is to evaluate if a topical ointment can cause
al eviation of pain. Thus, the study is powered to be able to show a 5 %
improvement in pain, measured on the Numeric Pain Intensity Scale, compared
to the positive control group (SD 50%). The computation indicates that 3,564
patients needed in each arm of the study to have 95%power to detect 5 %
improvement to be statistical y significant with a two-sided alpha error of p<0.01.
Thus a total of 10,692 patients are required, 3,564 in each of the three study
arms. Considering that up to 30% of the participants wil not complete the study
compliant with the protocol, participants wil be added for a total enrol ment of
6. SUBJECT RECRUITMENT
Study patients will be recruited by physicians who treat chronic pain conditions.
We expect to recruit up to 100 physician centers with each center contributing up
to 100 patients. Patients of any gender or race who meet the inclusion and
exclusion criteria are eligible for the study.
6.1. Inclusion Criteria:
1) 18 and 85 yr old female or male patients
2) Chronic extremity, joint muscular skeletal, neuropathy, or topical pain lasting
for more than 2 months, interfering with daily activities, work, or sleep.
3) Any systemic disease (cardiac, renal, or hepatic) must be wel control ed.
4) Absence of skin lesions at the site of application of the study medication
6.2. Exclusion Criteria:
1) Pregnant or lactating females or women at the child bearing potential not
2) Use of other topical or transdermal medication. Patients may be withdrawn for
a period of 2 weeks from all topical or transdermal medications prior to study
3) Hypersensitivity to local anesthetic or other ingredients of the compounded
4) Prior reconstruction skin surgery or skin grafts in the area of cream
application preventing absorption of cream.
5) Chronic pain conditions not expected to respond to topical pain medication
such as deep abdominal pain, or from conditions such as cancer, or gouty
6.3. Concomitant Treatment and Restrictions. Medication known to adversely
react with those in the topical ointments.
6.3.1. Concomitant Medications
Participants are required to avoid applying any analgesic topical cream or
transdermal medication for chronic pain conditions other than study medications.
Oral medications may have their dose reduced during the course of the study. A
reduction in oral medications wil be recorded in the patients chart and reported
on the electronic case report form. A reduction in oral medication dose wil be a
measure of efficacy of therapy if it correlates with no change, or a reduction of
pain as measured by the pain scale questions.
7. SCREENING FAILURES
Volunteers who are evaluated for entry into the study and fail to meet the
inclusion and exclusion criteria are defined as screening failures. A screening
log, which documents the screening number, volunteer’s initials, and reason for
screening failure, is to be maintained by the investigator. A copy of the log should
be retained in the investigator’s study files.
8. WITHDRAWAL FROM THE STUDY
Participants may withdraw from the study at any point. The Investigator shal
provide the reason for premature subject termination or withdrawal in the CRF.
9. STUDY METHODS 9.1. Pre-study screening
A brief evaluation if the patient meets inclusion and exclusion criteria wil be
performed and the result wil be documented in a Case Report Form. If the
volunteer is eligible for the study, he or she wil be al ocated to one of the two
study creams (at the prescribing physician’s discretion) or the control cream (if
patients insurance does not cover the formulated creams). The pre-study
screening must be performed within 2 weeks prior to study entry.
9.2. Study Procedures
If the volunteer meets the study entry criteria and has signed the informed
consent, the volunteer wil be scheduled for treatment.
Physical exam wil be performed by the physician at baseline 6 weeks of
Simple Descriptive Pain Intensity Scale and numeric pain intensity scale
will be employed at the baseline, after 6 weeks and 6 months of study.
9.2.1. Schedules of study topical cream administrations:
Types of ointment employed in this study:
Topical cream (active agent) – Type A or Type B
10. STUDY MATERIALS 10.1. Study Ointment 10.1.1. Topical creams wil be provided by the sponsor. 10.1.2. Voltaren will be purchased by the patients. 10.2. Medication Assessment: The physician at each site wil chose which
cream the patient shal receive (Cream A or B). If patients insurance does not
permit the compounded cream, then they wil be given the Voltaren Gel.
10.3. Case Report Forms (CRF)
Case report forms wil be prepared by American Institute Therapeutics an
11. PRECAUTIONS
Patients wil be closely monitored for potential side effects .
12. ADVERSE EVENTS
By definition, an adverse event (adverse experience) is any untoward, undesired,
unplanned clinical event in the form of signs, symptoms, disease, or laboratory or
physiological observations occurring in a human being participating in a clinical
study, regardless of causal relationship. This includes the fol owing:
• Any clinical y significant worsening of a pre-existing condition.
• an adverse event occurring from overdose, whether accidental or intentional
• an adverse event that has been associated with the discontinuation of the
All adverse reactions observed and/or reported during the course of the study
must be recorded in the patient’s Case Report Form (CRF). The Investigator
should record the type, severity (intensity), time of occurrence, duration, and
frequency (reoccurrence) of each event, and its relationship to the use of the
study medication (none, possibly related, probably related, definitely related).
The Investigator must record, when applicable, any treatment administered
because of any adverse reaction, inclusive of clinical observations, medications
used, laboratory procedures and/or other therapeutic measures applied. Any
adverse event must be recorded in the volunteer’s case report form.
13. STATISTICAL TESTING
The statistical analysis wil consist of computation of means and standard
deviations for continuous variables and frequencies for categorical variables.
Changes from baseline in continuous variables (i.e. pain intensity) fol owing
administration of topical cream wil be analyzed by paired sample t tests. If the
variables don’t have normal distribution, non-parametric tests wil be employed.
Categorical variables wil be evaluated by Fisher’s exact test. A two-sided alpha
error of p<0.05 wil be considered to be statistical y significant
14. MANAGEMENT OF EMERGENCIES 14.1. Acute Emergencies and Adverse Events
The subject wil be treated as medical y appropriate at the discretion of the
investigator. In case of a very unlikely adverse event that wil not spontaneously
resolve, or by the judgment of the investigator needs emergency treatment, the
volunteer wil be transferred to a hospital or emergency room for further care as
warranted. All emergencies and adverse events including treatments wil be
II. ADMINISTRATIVE SECTION This study does not include a drug needing FDA approval and therefore it is not by definition, a Clinical Drug Trial as defined by FDA. However, it will be conducted in accordance with FDA regulations to ensure integrity, participant’s safety and confidentiality, to meet moral standards of study conduction, and creditability of the results. 1. ETHICAL CONSIDERATIONS 1.1. Declaration of Helsinki
This study should be conducted in accordance with the Declaration of Helsinki
and laws of the State of Il inois and the United States of America that are
1.2. Institutional Review Board (IRB) or Independent Ethics Committee
Prior to starting the study, the Investigator wil submit a study agreement and
required information and the American Institute of Therapeutics wil obtain
1.3. Informed consent
Each participant must give informed consent prior to participating in the study.
Informed consent must be sought, and given freely, in conformity with applicable
Federal Law (The Code of Federal Regulations, 21 CFR Part 50). The study
should not begin until the document has been approved by the IRB. After the
informed consent form is signed, a copy wil be given to the participant and a
copy maintained on file in the participant’s confidential records, which wil remain
at the investigational site, and wil be available for inspection by Monitors from
IRB and/or the FDA, to the extent required by applicable law.
1.4. Obligation of the Investigator toward the I.R.B.
As part of his agreement to conduct the clinical study according to this protocol,
the Investigator wil provide assurance that any emergent problems, serious
adverse reactions, proposed protocol modifications and other matters that may
affect the status of the study wil be reported/submitted to the I.R.B. through the
1.5. Permission to Review Source Records
The investigator agrees that representatives of the IRB, Sponsor and, to the
extent required by applicable law, the FDA, or the Office of Human Research
Protections of the Department of Health and Human Services, wil have the right
to audit and review pertinent records relating to this trial. Permission to review
the study records wil be granted in each consent form signed by the participant.
2. PARTICIPANT IDENTIFICATION
The assignment of numbers for participant identification is based on the desire
for anonymity as wel as the requirement for randomization by number.
Participants should be identified only by their number, initials, age, and sex.
However, the investigator must maintain a participant’s log with names and
identifying information indicated above for a possible study audit.
3. MONITORING AND RECORDING OF STUDY DATA 3.1. Monitoring of Study
As an integral part of conducting this study, the Investigator understands and
accepts periodic monitoring visits by a duly identified and authorized representative
of the sponsor and an independent auditor. At each monitoring/auditing visit, the
Investigator and the monitor/auditor wil review the progress of the study and
compliance with the study protocol, discuss any emergent problems, and review the
CRFs for legibility, accuracy and completeness of the data.
3.2. Recording of Study Data
All study data must be recorded in the electronic CRFs provided by American
Institute Therapeutics according to the fol owing directions: Only the Investigator,
or an assistant duly authorized by the Investigator may make entries in the CRF.
The electronic CRF wil be kept and retained by American Institute
Therapeutics with required documentation appended to or part of each
CRF, for a period of not less than two (2) years after study completion.
Corrections to the CRFs: During the verification of the CRFs and data
entry procedures, errors and omissions may be identified that wil need to
be discussed with the Investigator. The Investigator agrees to comply with
4. ANALYSIS AND REPORTING OF STUDY DATA
The American Institute of Therapeutics wil col ect and analyze the study data
and provide a final report within 60 days of study completion to the IRB.
5. CONFIDENTIALITY AGREEMENT, USE OF DATA 5.1. Confidentiality of Disclosures
All documentation provided, inclusive of this protocol and the participant’s CRF is
considered confidential, and may not be disclosed by any other person without
the express written consent of the Institute and Sponsor. However, the
submission of this protocol and other necessary documentation to the
Investigator's IRB is expressly permitted under the terms of this protocol. Review
of al materials by the Sponsor, or Sponsor designated representative, or
consultants are also expressly permitted under the terms of this protocol.
6. AMENDMENTS TO THE PROTOCOL
No changes or amendments to this protocol may be made by the Investigator
after the protocol has been agreed to and signed by al parties unless such
change(s) or amendment(s) have been ful y discussed and agreed upon in
writing. Any change or amendment agreed upon wil be recorded in writing, the
written agreement wil be signed, and the signed agreement wil be appended to
[email protected] | www.ashleyfure.net | (b. 1982, USA) Harvard University – Cambridge, Massachusetts PhD Candidate in Music Composition, Expected Completion May 2013 Cursus 2, October 2010 – June 2011 Cursus 1, September 2008 – April 2009 Harvard University – Cambridge, Massachusetts Master of Arts in Music Composition with Distinction, June 2006 Bachelor of Music in
2ème cycle – MID – item 69 – Symptômes digestifs SYMPTOMES DIGESTIFS : I -Nausées, vomissements 40 % en phase terminale considérés comme * Étirement de la capsule hépatique* Irritation de la muqueuse intestinalemédiastinale (compression du défilé du X)STIMULATION DE LA ZONE GACHETTE DES CHEMOREC(Opiacés, Toxiques, Hypercalcémie, …) 2ème cycle
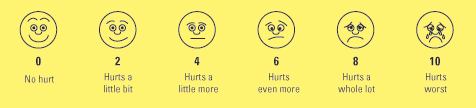
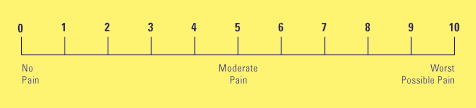 A simple descriptive pain intensity scale (Fig.1) and Numeric Rating Scale (Fig.2)
wil be employed as a supportive evidence for evaluating analgesic effect of the
4. DURATION
A simple descriptive pain intensity scale (Fig.1) and Numeric Rating Scale (Fig.2)
wil be employed as a supportive evidence for evaluating analgesic effect of the
4. DURATION