La spécificité du tadalafil est liée à sa longue demi-vie, permettant une action qui excède largement celle des autres inhibiteurs de PDE5. L’absorption digestive est complète, avec un pic plasmatique atteint en 2 heures environ. Le métabolisme est réalisé via CYP3A4, produisant des métabolites inactifs éliminés principalement dans les fèces. La sélectivité enzymatique est élevée, réduisant les effets indésirables extra-caverneux. Les réactions indésirables fréquentes incluent céphalées, bouffées vasomotrices et troubles digestifs légers. L’activité pharmacologique est stable, indépendamment de l’ingestion d’aliments. Dans les comparaisons de longue durée, acheter cialis pas cher est mentionné en relation avec les études portant sur la persistance d’efficacité et la constance de la cinétique plasmatique.
No job name
J. Am. Chem. Soc. 2000, 122, 12898-12900 Validation of a Model for the Complex of HIV-1 Reverse Transcriptase with Sustiva through Computation of Resistance Profiles
Robert C. Rizzo, De-Ping Wang, Julian Tirado-Rives, andWilliam L. Jorgensen*
Department of Chemistry, Yale UniVersityNew HaVen, Connecticut 06520-8107ReVised Manuscript ReceiVed October 24, 2000
All retroviruses depend on a virally encoded reverse tran-
scriptase enzyme (RT) to convert viral RNA into DNA forsubsequent incorporation into the host cell genome.1 Drug-designefforts to arrest reverse transcription in HIV have led to the FDA
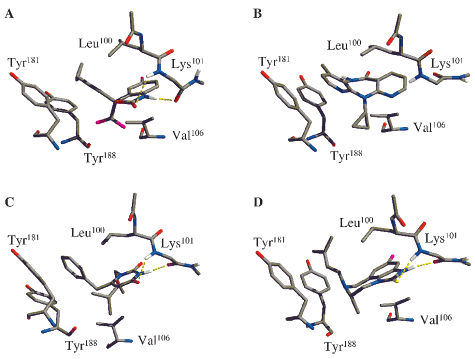
Figure 1. Orientation of the four NNRTIs in the HIVRT binding site.
approval of three non-nucleoside reverse transcriptase inhibitors
(A) Best docked structure of Sustiva. (B) Nevirapine from pdb entry 1vrt.
(NNRTIs), nevirapine, delavaridine, and efavirenz (Sustiva).
(C) MKC-442 from pdb entry 1rt1. (D) 9-Cl TIBO from pdb entry 1rev.
Additional compounds, including MKC-442, are in clinical trials(Table 1). Because of the low fidelity of HIVRT, the mutation
the X-ray and docked structures was 0.43-0.60 Å. These low
rate in the encoded proteins including HIVRT is great.2,3 As a
rmsd values and the limited flexibility of Sustiva are favorable
result, all HIVRT inhibitors incur resistance problems that
for the accuracy of the docking calculations.
adversely affect their clinical value.4,5 A measure of a drug’s
The best docked structure of Sustiva reveals that it makes
effectiveness against a mutation is given by the fold resistance,
interactions that are consistent with those for other NNRTIs and
which is the ratio of mutant to wild-type activities. Sustiva has
that it overlays well with the “butterfly” shape of nevirapine.
been shown to remain notably active against several common
Unlike nevirapine, hydrogen bonds are present between Sustiva
HIVRT point mutations including Val f Ala at position 106
and the protein backbone at position Lys101 that are similar to
(V106A) and Tyr f Cys at position 181 (Y181C) (Table 1).
those observed in the crystal structures with 9-Cl TIBO and MKC-
No HIVRT structure with Sustiva has been reported that may
442 (Figure 1). Nevirapine makes no formal ligand-protein
help explain its improved resistance profile. Herein, we have (a)
hydrogen bonds, but it does form a π-type hydrogen bond between
computed a structure for the Sustiva/HIVRT complex, (b)
the secondary amide hydrogen and Tyr18812 and water-mediated
validated the structure through computations of the effects of the
hydrogen bonds.10,12 The cyclopropyl ethynyl group of Sustiva
V106A and Y181C mutations on binding affinities for four drugs,
is positioned toward aromatic residues Tyr181 and Tyr188 in the
and (c) obtained structural insights on the improved effectiveness
same fashion as the methylpyridine fragment of nevirapine, the
of Sustiva. A binding site model6 was constructed from the 2.55-Å
benzyl ring of MKC-442, and the dimethylallyl group of 9-Cl
crystal structure of the MKC-442/HIVRT complex (pdb 1rt1)7
TIBO (Figure 1). Presumably, these aryl-π interactions all
with MKC-442 removed including only those residues within ∼15
contribute favorably to binding.4,5 Superposition based on the
Å of MKC-442. The MATADOR program8 was then used to dock
HIVRT CR atoms shows that these π fragments of the inhibitors
Sustiva into the NNRTI site.9 The other complexes were prepared
coincide spatially in the binding site and that Sustiva’s π fragment
analogously starting with coordinates from the X-ray structures
of nevirapine (pdb 1vrt),10 HEPT (pdb 1rti),7 and 9-Cl TIBO (pdb1rev)11 bound to HIVRT. The docking calculations placed Sustiva
An alternative binding mode suggested by Maga et al. was
in a reasonable position and orientation in the binding site in
based on an alignment of nevirapine and Sustiva in which the
comparison with the crystal structures for the complexes with
(9) MATADOR uses a Monte Carlo-based Tabu search algorithm. To keep
MKC-442, nevirapine, and 9-Cl TIBO (Figure 1). As controls,
the Tabu search focused on the known NNRTI binding site during the docking
MKC-442, nevirapine, 9-Cl TIBO, and HEPT were also docked
runs, a 50 kcal/mol Å2 half-harmonic restraining force was applied if the
back into their binding sites to verify that the docking protocol
distance between the ligand and the binding site center was greater than 5 Å. The defined binding site was roughly centered on the alkyne group of Sustiva.
could reproduce experimental structures. The lowest-energy
The Tabu list was set to be 25 and constructed from unique structures
structure generated during the docking runs was taken as the
considering energetic as well as geometric criteria. In total, 100 Tabu cycles
“best” structure and was found in all cases to reproduce closely
were performed with each Tabu search generating 100 randomly placed ligandpositions around the binding site. The decision to accept a new structure onto
the position and orientation observed in the crystal structures;
the Tabu lists is made after an intermolecular energy minimization and is
the rmsd for the non-hydrogen atoms of the four ligands between
based on both energetic and geometric criteria. The protein and ligand wererigid during the docking. The CM1P augmented OPLS-AA force field [Duffy,
(1) Katz, R. A.; Skalka, A. M. Annu. ReV. Biochem. 1994, 63, 133-173.
E. M.; Jorgensen, W. L. J. Am. Chem. Soc. 2000, 122, 2878-2888] provided
(2) Preston, B. D.; Poiesz, B. J.; Loeb, L. A. Science 1988, 242, 1168-
the initial structure of Sustiva; it was also used to determine the nonbonded
energies, which were stored on a spherical grid for efficiency. The total
(3) Roberts, J. D.; Bebenek, K.; Kunkel, T. A. Science 1988, 242, 1171-
intermolecular interactions between the ligand and protein amount to a measure
of both steric and electrostatic complimentarity; the lowest energy structure
(4) Tantillo, C.; Ding, J. P.; Jacobomolina, A.; Nanni, R. G.; Boyer, P. L.;
found during the simulations was taken as the “best” docked system. A
Hughes, S. H.; Pauwels, R.; Andries, K.; Janssen, P. A. J.; Arnold, E. J. Mol.
distance-dependent dielectric constant of 4 ( ) 4r) was used for all docking
Biol. 1994, 243, 369-387.
(5) De Clercq, E. AntiViral Res. 1998, 38, 153-179.
(10) Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.;
(6) Protein residues included in the binding site model were 91-110A,
Keeling, J.; Darby, G.; Jones, Y.; Stuart, D.; et al. Nat. Struct. Biol. 1995, 2,
161-205A, 222-242A, 316-321A, 343-349A, 381-383A, and 134-140B.
(7) Hopkins, A. L.; Ren, J. S.; Esnouf, R. M.; Willcox, B. E.; Jones, E.
(11) Ren, J.; Esnouf, R.; Hopkins, A.; Ross, C.; Jones, Y.; Stammers, D.;
Y.; Ross, C.; Miyasaka, T.; Walker, R. T.; Tanaka, H.; Stammers, D. K.;
Stuart, D. Structure 1995, 3, 915-26.
Stuart, D. I. J. Med. Chem. 1996, 39, 1589-1600.
(12) Rizzo, R. C.; Tirado-Rives, J.; Jorgensen, W. L. J. Med. Chem. 2000,
(8) Plount Price, M. L. Ph.D. Thesis, Yale University, 2000. J. Am. Chem. Soc., Vol. 122, No. 51, 2000 12899
Relative Free Energies of Binding (∆GFR) Estimated from Fold Resistance (FR) Valuesga Young, S. D.; Britcher, S. F.; Tran, L. O.; Payne, L. S.; Lumma, W. C.; Lyle, T. A.; Huff, J. R.; Anderson, P. S.; Olsen, D. B.; Carroll, S. S.;
Pettibone, D. J.; Obrien, J. A.; Ball, R. G.; Balani, S. K.; Lin, J. H.; Chen, I. W.; Schleif, W. A.; Sardana, V. V.; Long, W. J.; Byrnes, V. W.; Emini, E. A. Antimicrob. Agents Chemother. 1995, 39, 2602-2605. b Levin, J http://www.natap.org/reports/NR5-nnrti_update2.resis.htm. c Byrnes, V. W.; Sardana, V. V.; Schleif, W. A.; Condra, J. H.; Waterbury, J. A.; Wolfgang, J. A.; Long, W. J.; Schneider, C. L.; Schlabach, A. J.; Wolanski, B. S.; Graham, D. J.; Gotlib, L.; Rhodes, A.; Titus, D. L.; Roth, E.; Blahy, O. M.; Quintero, J. C.; Staszewski, S.; Emini, E. A. Antimicrob. Agents Chemother. 1993, 37, 1576-1579. d Balzarini, J.; Baba, M.; Declercq, E. Antimicrob. Agents Chemother. 1995, 39, 998-1002. e Baba, M.; Shigeta, S.; Yuasa, S.; Takashima, H.; Sekiya, K.; Ubasawa, M.; Tanaka, H.; Miyasaka, T.; Walker, R. T.; Declercq, E. Antimicrob. Agents Chemother. 1994, 38, 688-692. f Balzarini, J.; Karlsson, A.; Meichsner, C.; Paessens, A.; Riess, G.; Declercq, E.; Kleim, J. P. J. Virol. 1994, 68, 7986-7992. g FR ) mutant/wild-type activities, ∆G RT ln FR in kcal/mol. The columns show the structure, compound name, the assay type and reference
for the FR values, and ∆GFR for several common HIVRT mutations.
respectively, while ∆GA and ∆GB are the changes in free energyof binding for A and B with the mutant vs the wild-type protein. The quantities are related as ∆G
∆∆G, where ∆∆G is the experimentally observable differencein the fold resistance values, RT ln FR -
activity ratios from IC or EC values are expected to parallelbinding constant ratios for similar inhibitors.14 Computationally,one could mutate either the drug or the protein. However, we
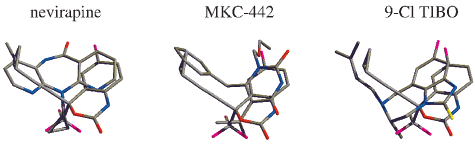
Figure 2. Overlays of the binding-site positions of nevirapine, MKC-
have chosen to perform the structurally simpler mutations of the
442, and 9-Cl TIBO with Sustiva in CPK colors.
protein; specifically, Val106 was mutated to Ala, and Tyr181 wasmutated to Cys in the presence of the four NNRTIs. Our resultsshould yield the observed experimental trends, if the proposedSustiva/HIVRT structure is correct.
Molecular dynamics (MD)15 equilibration and Monte Carlo16
free energy perturbation (MC/FEP)17 simulations were performedon the docked Sustiva structure and the equivalent nevirapine,MKC-442, and 9-Cl TIBO binding-site models, including 850water molecules in the FEP calculations. Aside from normalthermal oscillations, the positioning of Sustiva in the binding sitefrom the docking calculations was maintained in the MD andMC simulations. There is significant variability in the reportedfold resistance data, presumably due to the use of different assayconditions (Table 1). Sustiva, however, consistently emerges as
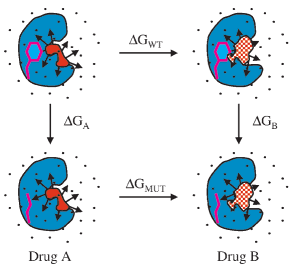
Figure 3. Thermodynamic cycle used to compute relatiVe fold resistance
more tolerant toward the Y181C and V106A mutations than the
values. The wild-type side-chain (magenta) is perturbed to the mutant
(14) Cheng, Y.; Prusoff, W. H. Biochem. Pharmacol. 1973, 22, 3099-
side chain in the presence of Drug A (solid red) and Drug B (checkered
red) while bound to a protein (cyan). Relative fold resistance ) ∆G -
(15) The docked Sustiva/HIVRT structure was subjected to a molecular
dynamics (MD) simulation to allow the HIVRT backbone and side chains torelax. The CM1P augmented OPLS-AA force field was used for all MDsimulations with the IMPACT program [Levy, R. M. et al., IMPACT Version
amide moiety of both drugs was superimposed.13 The present
c1.00; Schro¨dinger, Inc.: Jersey City, NJ, 1999]. Ten cycles of gradient-based
docking calculations did not find this orientation. Furthermore,
energy minimization were performed prior to the MD simulations, and the
forced placement of Sustiva in this alternative geometry yielded
complex was then restrained in the following manner. Protein residues wereallowed to move freely within ∼10 Å of the binding site (95-107A, 172A,
steric and electrostatic protein-ligand interaction energies ∼5 and
177-182A, 188-192A, 198A, 227A, 229A, 234-236A, 318-319A, 321A,
15 kcal/mol, respectively, less favorable than for our docked
and 135-139B). Movement was restrained for those residues in a 10-12 Å
structure. The alternative orientation is unlikely since the hydrogen
shell about the binding site, i.e., for residues 94A, 108A, 175-176A, 183A,187A, 225A, 237-239A, 317A, 320A, 349A, 382-383A, 134B, 140B with
bonds to the backbone of Lys101 would be sacrificed.
harmonic potentials. All other residues were restrained to their positions after
A computational experiment was then pursued to validate the
conjugate-gradient minimization. The Verlet algorithm was used to integrate
Sustiva model by predicting relative fold resistance values. The
Newton’s equations of motion using a time step of 0.001 ps and constanttemperature was maintained through coupling to a Berendsen temperature bath
methodology, which is a general computational approach to
using a relaxation parameter of 0.2 ps for the velocity scaling. Bond lengths
determining the impact of protein mutations on drug candidates,
were fixed by the SHAKE algorithm and a distance-dependent dielectric
hinges on the thermodynamic cycle in Figure 3. For two inhibitors,
) 4r) was used. First, 3 ps of initial equilibration was
performed at 100 K followed by 50 ps of equilibration at 300 K. Quenching
A and B, ∆GWT and ∆GMUT are the differences in free energy of
of the structure was performed by reducing the simulation temperature over
binding for B vs A with the wild-type and mutant proteins,
6 blocks of 4 ps each starting at 300 K and ending at 50 K. The same MDequilibration was also performed on the nevirapine, MKC-442, and the 9-Cl
(13) Maga, G.; Ubiali, D.; Salvetti, R.; Pregnolato, M.; Spadari, S.
TIBO structures, and the resultant complexes were then used in the MC
Antimicrob. Agents Chemother. 2000, 44, 1186-1194.
12900 J. Am. Chem. Soc., Vol. 122, No. 51, 2000
Relative Fold Resistance Energies (∆∆G) in kcal/mol for
be partly compensated by better alignment of the NH-O hydrogen
bond with Lys101 when the buttressing effect of the valine sidechain is reduced by conversion to alanine. In the MC simulations,
the hydrogen bond between the oxazinone NH of Sustiva and
the carbonyl oxygen of Lys101 is on average 0.1 Å shorter (1.77
vs 1.85 Å) when residue 106 is Ala rather than Val. The
3.88 ( 0.3 2.20, 2.71, 2.90 3.33 ( 0.4 2.34, 2.27, 2.95
interaction of the valine’s isopropyl group with the weakly
polarizable trifluoromethyl group is also likely less attractive than
9-Cl TIBO 3.01 ( 0.3 1.05, 0.41, 2.27 1.32 ( 0.5 0.66, 1.22, 1.48
the corresponding interactions with the cyclopropyl group of
a Values derived from Table 1.
nevirapine and the isopropyl and ethoxymethyl groups of MKC-442 (Figure 1). Thus, it is reasonable to propose, on the basis of
other drugs, especially nevirapine and MKC-442. Indeed, the
the present structure, that Sustiva’s improved resistance profile
present FEP results do predict Sustiva to be less affected by both
benefits from a combination of less favorable initial interactions
mutations than the other three inhibitors (Table 2). The agreement
with Tyr181 and Val106 and more favorable hydrogen bonding
of the computed free energies with the experimental results
with Lys101 in the V106A mutant. Consistently, the L100I
strongly supports the correctness of our docked model.
mutation is more damaging (Table 1) because Leu100 forms a
The structural model suggests some factors that render Sustiva
snug lid over the ring systems for all four inhibitors (Figure 1).
less affected by the Y181C and V106A mutations in comparison
Without adjustment, the branching at C rather than Cγ would
with the other compounds. It is well-known that the NNRTI
direct the methyl group of Ile100 directly into the rings. An
binding site is capable of accommodating structurally diverse
alternative strategy for improved resistance profiles is to enhance
inhibitors and that different inhibitors give rise to strikingly
interactions with immutable residues such as Trp229.18
different patterns of resistance mutations among ∼15 residues
In this work, we have presented a molecular model for the
that line the binding site.4,5 In general, this variability implies
important anti-HIV drug Sustiva bound to HIVRT. The resultant
that the effect of mutations on drug binding needs assessment on
structure reveals that Sustiva overlays well with the butterfly shape
a case-by-case basis. However, the Y181C mutant arises early
of nevirapine and makes similar contacts with HIVRT as do other
and confers resistance for many NNRTIs. This can be attributed
reported NNRTIs including hydrogen bonds with the backbone
to the loss of favorable aryl/π interactions, for example, between
of Lys101 (Figure 1). FEP methodology for the assessment of
the tyrosine and the methylpyridyl and benzyl rings of nevirapine
relative resistance profiles for drug candidates has been defined.
and MKC-442, and the dimethylallyl group of 9-Cl TIBO (Figure
Results from its application to four NNRTIs (Table 2) are in good
1). Loss of the interaction between Tyr181 and the smaller, less
agreement with the experimental activity trends and provide
polarizable cyclopropyl ethynyl group of Sustiva is expected to
evidence that the proposed binding mode for Sustiva is correct.
Sustiva’s relative insensitivity to the Y181C and V106A mutants
In the case of V106A, the residue is tucked under the benzene
appears to arise from a mix of relatively weaker interactions with
ring of Sustiva and is in van der Waals’ contact with the
Tyr181 and Val106 and improvement of hydrogen bonding for
trifluoromethyl group. Reduction of these interactions appears to
Ala106. These findings highlight the power of molecular modelingfor structure and binding affinity predictions and its potential for
(16) Each protein-inhibitor complex was briefly energy-minimized prior
to the Monte MC simulations using a distance-dependent dielectric constantof 4 ( ) 4r). The CM1P augmented OPLS-AA force field was used with the
Acknowledgment. This work was supported by the National Institute
MCPRO program [Jorgensen, W. L. MCPRO Version 1.65; Yale University:
of Allergy and Infectious Diseases (AI44616). We thank Melissa L. Plount
New Haven, CT, 2000]. For the MC simulations, a water cap with a 22-Åradius was used containing ∼850 TIP4P water molecules and the system was
Price for computational assistance with the docking calculations, and Dr.
partitioned into rigid residues (91-94A, 109-110A, 116-178A, 184-185A,
Marilyn B. Kroeger Smith, Professor Richard H. Smith, and Mark A.
192-197A, 199-205A, 222-224A, 230-232A, 240-242A, 316-317A,
320-321A, 343-349A, 381-383A, 134-135B, 137B, 140B) and flexibleresidues (95-108A, 179-183A, 186-191A, 198A, 225-229A, 233-239A,
Note Added in Proof. A crystal structure has now appeared
318-319A, 136B, 138B). All HIVRT side chains within ∼10 Å from the
center of the water cap were sampled, the protein backbone was fixed, and
for a Sustiva/HIVRT complex [Ren, J.; Milton, J.; Weaver, K.
each inhibitor was fully flexible. Each solvated complex was subjected to 1
L; Short, S. A.; Stuart, D. I.; Stammers, D. K. Structure 2000, 8,
million configurations of solvent-only equilibration, 10 million of equilibration,
1089-1094]. It fully confirms the correctness of the structure
and 10 million configurations of averaging per window during the FEPsimulations. For additional MC simulation parameters and protocols, see
(17) For a recent discussion of FEP methodology, see: Jorgensen, W. L.
Free Energy Changes in Solution. In Encyclopedia of ComputationalChemistry; Schleyer, P. v. R., Ed.; Wiley: New York, 1998; Vol. 2, pp 1061-
(18) Hopkins, A. L.; Ren, J.; Tanaka, H.; Baba, M.; Okamato, M.; Stuart,
D. I.; Stammers, D. K. J. Med. Chem. 1999, 42, 4500-4505.
USA Gymnastics Online: Technique: Approaches to Treating ADHD Alternative Medical Approaches to Treating Attention Deficit/Hyperactivity Disorder By Larry Nassar, D.O., A.T.C., USA Gymnastics National Medical Coordinator Michigan State University, College of Osteopathic Medicine, Assistant Professor, Department of Family & Community Medicine Attention Deficit Hyperactivity Disorder (ADHD
Article: ““Interpose Your Friendly Hand”: Political Supports for the Exercise of Judicial Review by the United States Supreme Court” Author: Keith E. Whittington Issue: November 2005 Journal : American Political Science Review This journal is published by the American Political Science Association. All rights reserved. APSA is posting this article for public view on its
 J. Am. Chem. Soc. 2000, 122, 12898-12900
J. Am. Chem. Soc. 2000, 122, 12898-12900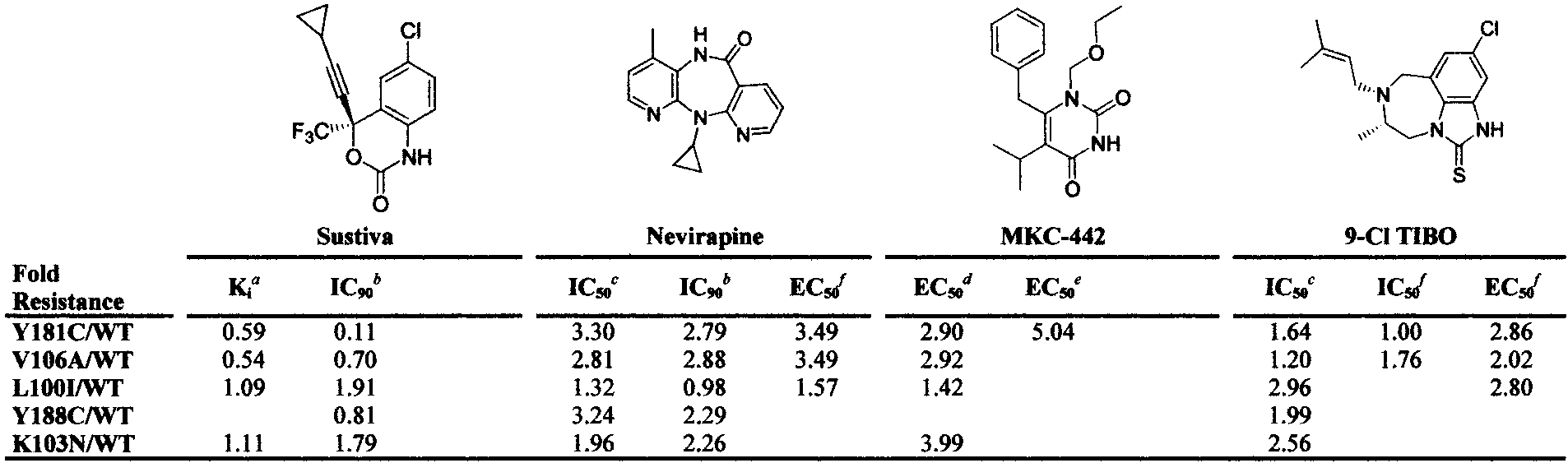
 J. Am. Chem. Soc., Vol. 122, No. 51, 2000 12899
Relative Free Energies of Binding (∆GFR) Estimated from Fold Resistance (FR) Valuesg
a Young, S. D.; Britcher, S. F.; Tran, L. O.; Payne, L. S.; Lumma, W. C.; Lyle, T. A.; Huff, J. R.; Anderson, P. S.; Olsen, D. B.; Carroll, S. S.;
Pettibone, D. J.; Obrien, J. A.; Ball, R. G.; Balani, S. K.; Lin, J. H.; Chen, I. W.; Schleif, W. A.; Sardana, V. V.; Long, W. J.; Byrnes, V. W.; Emini,
J. Am. Chem. Soc., Vol. 122, No. 51, 2000 12899
Relative Free Energies of Binding (∆GFR) Estimated from Fold Resistance (FR) Valuesg
a Young, S. D.; Britcher, S. F.; Tran, L. O.; Payne, L. S.; Lumma, W. C.; Lyle, T. A.; Huff, J. R.; Anderson, P. S.; Olsen, D. B.; Carroll, S. S.;
Pettibone, D. J.; Obrien, J. A.; Ball, R. G.; Balani, S. K.; Lin, J. H.; Chen, I. W.; Schleif, W. A.; Sardana, V. V.; Long, W. J.; Byrnes, V. W.; Emini,