J Mol Model (2001) 7:26–33DOI 10.1007/s008940100013
Somsak Tonmunphean · Vudhichai Parasuk Sirirat Kokpol
Automated calculation of docking of artemisinin to heme
Received: 28 September 2000 / Accepted: 1 February 2001 / Published online: 4 April 2001 Springer-Verlag 2001
Abstract We report automated molecular docking of ar-
might be indicative of a different mode of action from
temisinin to heme. The effects of atomic charges, and li-
those of the other antimalarial drugs, and hence the high
gand and heme structures on the docking results were in-
potency against the resistant strains. Although the mech-
vestigated. Several charge schemes for both artemisinin
anism of its antimalarial activity is still in doubt, there is
and heme, artemisinin structures taken from various opti-
general agreement on the significance of the endoperox-
mization methods and X-ray data, and five heme models,
ide group of artemisinin to the antimalarial activity. This
were employed for this purpose. The docking showed
is evident from the inactivity of the deoxyartemisinin
that artemisinin approaches heme by pointing O1 at the
compound that lacks the endoperoxide moiety [7]. In ad-
endoperoxide linkage toward the iron center, a mecha-
dition, in-vitro experiments revealed that iron is required
nism that is controlled by steric hindrance. This result
for artemisinin to have antimalarial activity [8, 9, 10].
differs from that reported by Shukla et al. which suggest-
In humans, malarial parasites digest more than 70%
ed that heme binds with artemisinin at the O2 position.
of the hemoglobin within the infected red blood
The docking results also depended on the structures of
cell [11], giving globin and heme as the products. The
both artemisinin and heme. Moreover, the atomic
globin is hydrolyzed to give amino acids, which are used
charges of heme have a significant effect on the docking
in protein synthesis by the parasite. The toxic heme
(Fig. 2) is mostly detoxified by a specific mechanism ofheme polymerization into hemozoin. The heme polymer-
Keywords Docking · Antimalarial drug · Endoperoxide ·
ization is a target for some antimalarials, such as chloro-
quine, that inhibit this process [12]. A recent study re-ported that artemisinin also inhibits heme polymeriza-tion [13]. The chloroquine-resistant strain of Plasmodi-um berghei that lacks hemozoin, possibly because hemepolymerization does not occur, is also resistant to arte-
Malaria is one of the most widespread and prevalent en-
misinin [14]. This supports the view that inhibition of
demic diseases; it threatens approximately 40 percent of
heme polymerization is the mode of action of artemis-
the world's population in more than 90 countries. This
inin. It is very possible that artemisinin interacts with
disease is estimated to cause approximately 300 to 500
heme and hence inhibits the polymerization process.
million illnesses and up to 3 million deaths each year [1].
It has been proposed that heme iron attacks the endo-
This tremendous prevalence might be partly because of
peroxide linkage of artemisinin either at the O1 [15] or
the resistance of malaria parasites to most antimalarialagents, e.g. chloroquine, quinine, and mefloquine [2, 3]. Artemisinin (Fig. 1), a sesquiterpene endoperoxide iso-
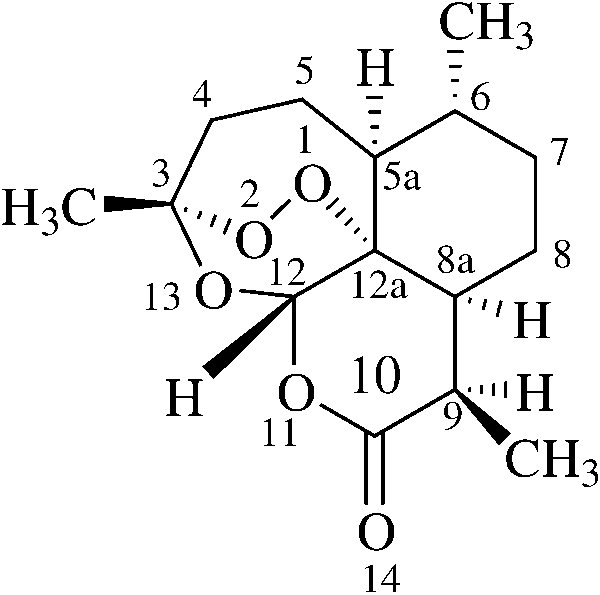
Fig. 1 The structure of arte-
lated from a Chinese medicinal herb [4], is, however, a
potent antimalarial drug against the resistant strains ofPlasmodium falciparum [5, 6]. Its unusual structure
S. Tonmunphean · V. Parasuk (✉) · S. Kokpol
Department of Chemistry, Faculty of Science, Chulalongkorn University, Patumwan, Bangkok, 10330 Thailande-mail: [email protected].: 662 218 5221, Fax: 662 252 1730
O2 position [16] (Fig. 3). In pathway A, heme iron at-
results obtained will give information on how the chemi-
tacks the compound at the O2 position and produces a
cal structure of the drug should be modified to achieve
free radical at the O1 position. Later it rearranges to
suitable interactions. Hence, this could bring about the de-
form the C4 free radical. In pathway B, heme iron at-
velopment of new and more effective drugs.
tacks the compound at the O1 position and produces a
For this reason, Shukla and co-workers [17] studied
free radical at the O2 position. After that the C3–C4
the docking of artemisinin and deoxyartemisinin with
bond is cleaved to give a carbon radical at C4. It has
hemin [Fe(II) and Fe(III)] using the Dock module in
been suggested that the C4 free radical in both pathways
SYBYL software, a direct docking algorithm. In their
is an important substance in antimalarial activity [10].
study the artemisinin structure was built from the crys-
The mechanism of action of any drug is very important
tallographic X-ray structure of artemether. Although the
in drug development. Generally, the drug compound binds
study did not elaborate on how the structure of artemis-
with a specific target, a receptor, to mediate its effects.
inin was obtained from artemether, it is very likely that
Therefore, suitable drug–receptor interactions are required
the geometry optimization was performed at either the
for high activity. Understanding the nature of these inter-
molecular mechanics or semi-empirical level, because
actions is very significant and theoretical calculations, in
only these methods are available in SYBYL. For their
particular the molecular docking method, seem to be a
docking calculations, only three orientations of artemis-
proper tool for gaining such understanding. The docking
inin around the hemin molecule were considered. Fur-thermore, the Gasteiger method, an empirical methodimplemented in the SYBYL, was used for the atomiccharge calculations. Because this empirical method hasno parameters for iron, however, the charge of the hemeiron was assigned under the assumption that the changein the charge distribution of the heme iron should beequal to that of the heme model where iron was replacedby aluminum. Moreover, the general parameters for met-als were used in the docking calculations. The dockingscheme they employed might influence the docking re-sult in favor of one of the heme–artemisinin configura-tions and yield an inaccurate model for the complex. It isquite important to have an accurate model for theheme–artemisinin complex, because this knowledge canbe used to design better and more potent antimalarial
Fig. 2 The structure of heme Fig. 3 Proposed mechanism of action of artemisinin
In this study, automated docking calculations were
each docking calculation and 100 docking calculations
performed to eliminate the bias in selecting preferred
were performed. A cluster analysis was used to catego-
configurations (orientations). Thus, all possible configu-
rize all 100 docked configurations into groups. Configu-
rations between heme and artemisinin were explored.
rations with root-mean-square-deviation (rmsd) values of
The crystallographic X-ray structure of artemisinin was
less than 1 Å were grouped together. The lowest energy
used for artemisinin instead of that of artemether, which
configuration was selected as a representative for each
is quite different from the artemisinin structure, especial-
group. Our attention was focused on the group with the
ly at the lactone ring. In addition, because few crystallo-
highest number of members, referred to as “the most oc-
graphic X-ray structures of artemisinin derivatives are
curring configuration”. Thus, it is most probable that this
available, it is worth establishing a suitable geometry op-
configuration represents the real system.
timization scheme to determine structures of artemisininderivatives for further investigations [18]. For the hemeiron, accurate ab initio calculations were performed to
obtain its atomic charge (and those of artemisinin) in-stead of using a crude approximation for the charge of
In addition to the crystallographic X-ray structure, the
iron, and specific parameters for iron were used in the
docking of heme and the optimized geometries of arte-
docking calculations. The effects of different heme struc-
misinin obtained at AM1, HF/3-21G, and HF/6-31G*
tures were also considered. Thus, five heme structures
levels of theory were investigated (these structures were
taken from the literature were studied.
taken from Ref. [24]). For the receptor molecule, five
The knowledge obtained from this study has been
heme structures, i.e., heme-pdb, heme-model, heme-
used as a guide for series of docking experiments be-
hemin, heme-deoxy, and heme-oxy, were considered.
tween heme and artemisinin derivatives and we found a
These structures are all different owing to the source of
very pronounced relationship between their binding en-
heme and the oxidation state of iron. The first structure,
ergies and antimalarial activity [18].
heme-pdb, was taken from the Protein Data Bank (id1CTJ) [25]. In this structure, Fe positions itself slightlyabove the porphyrin plane (Fig. 4a). The second struc-
ture, heme-model, which was taken from the AMBERdatabase [26] has the planar geometry (Fig. 4b). The
third structure, heme-hemin, was modified from thecrystallographic X-ray structure of chlorohemin of the
AutoDock 2.4 [19], an automated docking program, was
Cambridge Crystallography Data Bank [27]. This struc-
used for the docking calculations. The automated docking
ture has a pyramidal shape with Fe on the top (Fig. 4c).
is performed using a simulated annealing Monte Carlo
In the process of hemoglobin degradation by the ma-
simulation in combination with a rapid grid-based ener-
laria parasite, the proximal ligand may possibly still be
gy-evaluation method. A grid map of dimensions
attached to the heme iron and, therefore, it is very possi-
25×25×25 Å3 with a 0.5 Å spacing was selected. The
ble that the histidine remains with the heme structure. As
combined AMBER/MMFF parameters [20, 21] were
a result, the fourth and the fifth structures, heme-deoxy
chosen for the Lennard–Jones 12,6 potentials and Cou-
and heme-oxy, respectively, were obtained from the
lomb potentials to calculate the interaction energy, in-
modifications of deoxy and oxy forms of hemoglobin
stead of using the AMBER force field that contains no
which contain histidine as the proximal. Both deoxy and
parameters for iron. These parameters were taken from
oxy forms of hemoglobin were taken from the Protein
Data Bank (id 1A3 N and 1HHO, respectively). In the
In one docking calculation, the simulations were per-
heme-deoxy, the histidine pulls the Fe atom to lie below
formed for 100 annealing cycles. At the first cycle, the ini-
the protoporphyrin plane and gives it a basin-like struc-
tial annealing temperature (RT) was set to 100 kcal mol–1
ture (Fig. 4d). In the oxy hemoglobin structure, there are
and then the temperature was reduced at the rate of 0.90
six coordinations for heme iron, i.e. with four N atoms in
per cycle. During each cycle, the ligand was gradually
the protoporphyrin ring, with the proximal ligand (histi-
moved by a random displacement with a maximum trans-
dine), and with O . Thus, for docking purposes, the O
lation step of 0.2 Å and a maximum orientation step of 5°.
coordination was deleted while maintaining the coordi-
The energy of the new configuration was then calculated.
nates of the rest; this modified structure was taken as the
The selection of the new configuration was based on the
receptor structure. As in heme-deoxy, the protoporphyrin
Metropolis algorithm [23]. The cycle terminates if the li-
plane has a basin-like structure, because of the attraction
gand makes 30,000 accepted or 30,000 rejected moves.
to the heme iron by histidine. Interaction with O causes
Then the simulation moves to the next cycle.
the Fe atom to be drawn up above the plane (Fig. 4e),
Because the Monte Carlo simulation is based on ran-
however, and thus results in a structure which is marked-
dom movements, the final docked configuration depends
on the starting configuration. To avoid any bias and togenerate as many final docked configurations as possi-ble, the starting configuration was assigned randomly for
In docking calculations, the electrostatic potential is builtfrom atomic charges. Therefore, the choices for atomiccharges of both the ligand and receptor would have an effect on the docking results. Using charges obtained from ZINDO/S, HF/STO-3G, HF/3-21G, andHF/6-311G** levels of theory for heme-pdb, the dockingto the artemisinin X-ray structure with HF/3-21G
Fig. 4 The structures of the five heme compounds: (a) heme-pdb,
charges was performed. The results in Table 1 showed
(b) heme-model, (c) heme-hemin, (d) heme-deoxy, (e) heme-oxy
that the docking configurations depend on the heme-pdbatomic charges and especially the charge of Fe. With theexception of ZINDO/S charges, all docking calculations
agree that the heme iron binds with endoperoxide oxy-gens, where the O1–Fe distance is the shortest. Among
To investigate the effect of the atomic charge on docked
these calculations, docking with HF/6-311G** charges
configurations, atomic charges of both artemisinin and
yielded the shortest O1–Fe distance of 2.51 Å. This
heme obtained at various levels of theory were used. O1–Fe distance is markedly much shorter than those pre-For heme, the ZINDO/S, STO-3G, HF/3-21G, and dicted using HF/STO-3G (2.71 Å) and HF/3-21GHF/6-311G** atomic charges were calculated. For (2.70 Å) charges. For the binding energy, the dockingartemisinin, atomic charge calculations were performed
with HF/STO-3G charges gave the lowest energy while
at AM1, PM3, HF/3-21G, HF/D95, HF/6-31G*, and that with HF/6-311G** charges gave the second lowest. HF/6-311G**. All quantum chemical calculations were
Thus, the employed charge scheme for heme does have a
carried out using the Gaussian 94 program [28].
profound effect on the docking result. It is, however,quite difficult to judge which charge scheme leads to themost accurate result, because there is no supporting ex-
Table 1 Results for docking of heme-pdb with different atomic charges and the artemisinin X-ray structure with HF/3-21G charge
a The underlined values are the shortest O–Fe distances
Table 2 Results for docking of heme-pdb with HF/6-311G** charge and the artemisinin X-ray structure with different atomic charges
a The underlined values are the shortest O–Fe distances
Table 3 Atomic charges of
perimental evidence. Theoretically, HF/6-311G** is the
HF/3-21G, and HF/6-31G**. Comparison of these opti-
most accurate level of theory employed. It is, therefore,
mized geometries with the crystallographic X-ray struc-
reasonable to choose atomic charges from HF/6-311G**
ture [29] showed that HF/3-21G gave geometry parame-
for heme in further docking calculations. To study the ef-
ters in good agreement with those of crystallographic X-
fect of atomic charges of artemisinin, the docking calcu-
ray data, especially for the bond length of the endoperox-
lations using various charge schemes, i.e., AM1, PM3,
ide linkage, whereas AM1 and HF/6-31G* yielded an
HF/3-21G, HF/D95, HF/6-31G*, and HF/6-311G** for
O–O bond distance that was too short. This shorter O–O
the artemisinin X-ray structure and HF/6-311G**
bond length for AM1 and HF/6-31G* is not only found in
charges for heme-pdb structure were performed. The
artemisinin but also in other peroxide systems [30]. The
docking results are given in Table 2 and the atomic
HF/3-21G method is, therefore, recommended for the op-
charges of four oxygen atoms in artemisinin for each
timization of artemisinin derivatives. This recommenda-
charge scheme are listed in Table 3. From Table 2, the
tion is, however, based on geometrical criteria only, which
dockings with ab initio charges (HF/3-21G, HF/D95,
does not necessarily guarantee good docking results.
HF/6-31G*, and HF/6-311G**) gave similar results,
To validate the use of this optimized artemisinin
whereas those with semi-empirical charges (AM1 and
structure, the docking calculations between heme-pdb
PM3) gave longer O–Fe distances. Thus, for the sake of
with HF/6-311G** atomic charges and the AM1, HF/3-
saving CPU times, the HF/3-21G charges were chosen
21G, and HF/6-31G* optimized structures of artemisinin
were performed. The results were compared with thoseobtained using the artemisinin crystallographic X-raystructure. For the optimized structures, atomic charges of
artemisinin were taken according to the optimizationmethods, i.e. AM1 charges for the AM1 structure, etc.
In our previous study [24], artemisinin was geometry-opti-
For the X-ray structure, three docking calculations using
mized at various levels of accuracy, ranging from the
AM1, HF/3-21G, and HF/6-31G* charges for artemis-
semi-empirical CNDO and AM1 to ab initio HF/STO-3G,
inin were performed. The docking results are given in
Table 4 Results for docking of heme-pdb with HF/6-311G** charge and artemisinin optimized structures at various levels of theory
a The underlined values are the shortest O–Fe distances
Table 5 Results for docking of different heme structures and artemisinin HF/3-21G optimized structure
a The underlined values are the shortest O–Fe distances
Table 4. Comparison of the configurations which occur
most often reveals good agreement between the dockingusing the X-ray structure and HF/3-21G structure for ar-
To investigate the effect of the heme structure, five heme
temisinin. The largest deviation is 0.03 Å (O11–Fe dis-
structures were selected as described in the section on
tance). Comparing the AM1 and the X-ray structures, the
computational details. The atomic charges were assigned
optimized structure yielded an O1–Fe distance that was
as HF/6-311G** charges for all five heme molecules.
short by 0.2 Å, with the largest deviation 0.55 Å (O2–Fe
For artemisinin compounds, the HF/3-21G optimized
distance). Although much better for docking than the
structure and atomic charges were used. The results are
AM1 structure, when comparing the HF/6-31G* and X-
ray structures, the optimized structure gave an O1–Fe
The heme structure chosen does have an effect on the
distance that was too long by 0.08 Å, with the largest de-
docking results. Although we could not observe agree-
viation of 0.12 Å (O11–Fe distance). The discrepancy
ment on O–Fe distances, all docking calculations with
between the docking results obtained from the AM1 and
different heme structures (except heme-deoxy) suggested
the HF/6-31G* structures and the X-ray structure is
that artemisinin prefers to dock at endoperoxide oxygens
clearly rooted in the deficiency of the methods, which
(O1 and O2). Using heme-pdb for the heme structure,
yielded O–O distances that were too short. Thus, the
the docking results showed that artemisinin pointed its
method which gives a good structure (compared with the
endoperoxide moiety toward the heme iron for the most
X-ray structure) will also give good docking results.
occurring configuration. The O1–Fe and O2–Fe distanc-
HF/3-21G is, therefore, the recommended method for
es of were measured and found to be 2.49 Å and 3.12 Å,
geometry optimization of artemisinin derivatives in fur-
respectively (Fig. 5a); the binding energy obtained was
ther study although it has a lower level of accuracy than
–31.40 kcal mol–1. Owing to the planar structure of the
HF/6-31G*. It can be argued that for artemisinin deriva-
heme-model, the repulsion between artemisinin and the
tives it is possible that the good agreement between the
protoporphyrin ring of heme prevents artemisinin from
HF/3-21G and the X-ray structures no longer exists, so it
approaching the heme iron as closely as for heme-pdb.
would be wiser to employ the more accurate method,
Thus, the O1–Fe and O2–Fe distances of 2.75 Å and
HF/6-31G*. From previous calculations on artemisinin,
3.66 Å (Fig. 5b) were obtained, with a binding energy of
however, and the current docking results the difference
–29.92 kcal mol–1, the weakest among the heme struc-
between the structures obtained from the two methods is
tures investigated. Unlike the first two models, the dis-
not pronounced. Thus, the HF/3-21G method is still pre-
tances between the endoperoxide oxygens and Fe for
ferred, because of its faster computation time.
heme-hemin are very short, 2.00 Å and 2.65 Å forO1–Fe and O2–Fe (Fig. 5c), with a binding energy of–33.13 kcal mol–1 (the lowest). This is probably becauseof the pyramidal-like structure of heme-hemin which fa-cilitates the approach of Fe to the endoperoxide moiety.
most occurring configuration, which has the shorterO13–Fe distance of 3.26 Å, compared with 5.95 and5.53 Å for O1–Fe and O2–Fe (Fig. 5d). Interestingly, thesecond most occurring configuration has shorter O1–Feand O2–Fe distances. Still, this distance is longer thanthose obtained from the docking with other heme struc-tures. For heme-oxy, the most occurring configurationhas O1–Fe as the shortest heme–artemisinin distancewith the binding energy of –32.32 kcal mol–1 (Fig. 5e). The O1–Fe and O2–Fe distances of 2.52 Å and 3.32 Åare comparable with those of heme-pdb. Note that heme-oxy and heme-pdb have similar structures.
From the results from the five heme structures, it can
Fig. 5 Docking configuration between artemisinin and (a) heme-pdb,
be concluded that the structure of the heme molecule has a
(b) heme-model, (c) heme-hemin, (d) heme-deoxy, (e) heme-oxy
significant effect on the docking configurations. The sterichindrance at the Fe position plays an important role in thebinding. The proximal ligand that increases the steric hin-
The O1–Fe distance of 2.00 Å is comparable with the
drance at the Fe position would significantly affect the
experimental bond length between the heme iron and
docking results, as in heme-deoxy. If, however, the proxi-
oxygen atom in oxyhemoglobin A (1.86 Å), taken from
mal ligand does not increase the steric hindrance, results
similar to those without the proximal ligand, i.e. for heme-
For the heme-deoxy, because of its basin-like oxy and heme-pdb, would be obtained. Therefore, the
structure (see Fig. 4d), the binding with the endo-
heme structures which facilitate binding between Fe and
peroxide moiety of artemisinin is less favorable and a
endoperoxide oxygens, such as heme-pdb, heme-hemin,
stronger O13–Fe attraction is resulted (binding energy
and heme-oxy, are recommended for further investigation
–31.03 kcal mol–1). This could be observed from the
All docking calculations similarly reported O1–Fe as
the shortest heme–artemisinin distance and O2–Fe as thesecond shortest. It could then be concluded that iron in
1. World Health Organization. The World Health Report 1999.
heme interacts with O1 more preferably than O2, a pref-
2. Moore, D. V.; Lanier, J. E. Am. J. Trop. Med. Hyg. 1961, 10, 5. 3. Mockenhaupt, Parasitol. Today 1995, 11, 248.
erence which might arise from the more negative charge
4. Qinghaosu Antimalaria Coordinating Research Group Chin.
at O1 and the steric hindrance at O2. This observation is
Med. J. 1979, 92, 811.
in agreement with the proposal of Posner et al. [16]
5. Klayman, D. L. Science 1985, 228, 1049.
(pathway B). From their docking results, however,
6. Luo, X. D.; Shen, C.-C. Med. Res. Rev. 1987, 7, 29. 7. China Cooperative Research Group on Qinghaosu and Its
Shukla et al. [17] reported O2–Fe as the shortest
Derivatives as Antimalarials J. Trad. Chin. Med. 1982, 2, 3.
heme–artemisinin distance. This disagreement is possi-
8. Meshnick, S. R.; Thomas, A.; Ranz, A.; Xu, C.-M.; Pan, H.-Z.
bly the result of using poor atomic charges, ab initio
Mol. Biochem. Parasitol. 1991, 49, 181.
rather than empirical models, and a poor geometry for
9. Meshnick, S. R.; Yang, Y.-Z.; Lima, V.; Kuypers, F.;
Kamchonwongpaisan, S.; Yuthavong, Y. Antimicrob. Agents Chemother. 1993, 37, 1108.
10. Posner, G. H.; Oh, C. H.; Wang, D.; Gerena, L.; Milhous, W.
K.; Meshnick, S. R.; Asawamahasakda, W. J. Med. Chem.1994, 37, 1256.
11. Francis, S. E.; Sullivan, D. J.; Goldberg, D. E. Annu. Rev. Microbiol. 1997, 51, 97.
The docking results for five heme structures all agreed
12. Slater, A. F. Pharmacol. Ther. 1993, 57, 203.
that the heme iron approaches the endoperoxide moiety
13. Pandey, A. V.; Tekwani, B. L.; Singh, R. L.; Chauhan, V. S. J.
at the O1 position in preference to the O2 position. The
Biol. Chem. 1999, 274, 19383.
docking configuration depends on the structures and
14. Peters, W.; Li, Z. L.; Robinson, B. L.; Warhurst, D. C. Ann.Trop. Med. Parasitol. 1986, 80, 483.
atomic charges of both artemisinin and heme. The 15. Jefford, C. W.; Vicente, M. G. H.; Jacquier, Y.; Favarger, F.;
HF/3-21G level of theory is suitable for the geometry
Mareda, J.; Millasson Schmidt, P.; Brunner, G.; Burger, U.
optimization of artemisinin and its derivatives. The
Helv. Chim. Acta 1996, 79, 1475.
docking configurations were significantly affected by
16. Posner, G. H.; Cummings, J. N.; Ploypradith, P.; Oh. C. H. J.
the atomic charges of heme and to a much lesser extent
Am. Chem. Soc. 1995, 117, 5885.
17. Shukla, K. L.; Gund, T. M.; Meshnick, S. R. J. Mol. Graph.
by the atomic charges of artemisinin. The high quality
1995, 13, 215.
atomic charges, 6-311G**, are recommended for the
18. Tonmunphean, S.; Parasuk, V.; Kokpol S. Quant. Struct–Act.
electrostatic potential of heme. Heme structures with no
Rel. 2000, 19, 475.
or little steric hindrance at the Fe position facilitate
19. Morris, G. M.; Goodsell, D. S.; Huey, R.; Olson, A. J., Auto-
Dock version 2.4, The Scripps Research Institute, Department
binding of heme and endoperoxide oxygens as in heme-
of Molecular Biology, MB-5, La Jolla, California, U.S.A.
pdb, heme-hemin, and heme-oxy, and they are recom-
20. Weiner, S. J.; Kollman, P. A.; Nguyen, D. T.; Case, D. A. J.
mended for use in docking calculations. Comparison of
Comput. Chem. 1986, 7, 230.
docking results for heme-deoxy and heme-oxy, the
21. Halgren, T. A. J. Am. Chem. Soc. 1992, 114, 7827. 22. http://www.scripps.edu/pub/olson-web/doc/autoflex/parame-
heme-oxy structure, whose structure is very close to the
receptor structure in the bound state, gave docking re-
23. Morris, G. M.; Goodsell, D. S.; Huey, R.; Olson, A. J. J. Com-
sults that are in agreement with those of other heme
put-Aided Mol. Des. 1996, 10, 293.
24. Tonmunphean, S.; Kokpol, S.; Parasuk, V.; Wolschann, P.;
Winger, R. H.; Liedl, K. R.; Rode, B. M. J. Comput-AidedAcknowledgements The authors would like to thank the Comput- Mol. Des. 1998, 12, 397.
er Center of the University of Vienna, Austria, for providing com-
puting resources at their workstation clusters and the Austrian-
26. Case, D. A.; Pearlman, D. A.; Caldwell, J. W.; Cheatham, T.
Thai Center (ATC) for Computer-Assisted Chemical Education
E. III; Ross, W. S.; Simmerling, C. L.; Darden, T. A.; Merz, K.
and Research, Department of Chemistry, Faculty of Science,
M.; Stanton, R. V.; Cheng, A. L.; Vincent, J. J.; Crowley, M.;
Chulalongkorn University, Thailand, for computer resources and
Ferguson, D. M.; Radmer, R. J.; Seibel, G. L.; Singh, U. C.;
other facilities. Tonmunphean S. would like to thank Prof. Dr. Peter
Weiner, P. K.; Kollman, P. A. (1997), AMBER 5, University of
Wolschann, Institute of Theoretical Chemistry and Molecular
Biology, University of Vienna, Austria for helpful discussion and
28. Gaussian 94, Revision B.3, M. J. Frisch, G. W. Trucks, H. B.
Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Petersson, J. A. Montgomery, K. Raghavachari, M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz,J. B. Foresman, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart, M. Head-Gordon, C. Gonzalez, and J. A. Pople,Gaussian, Inc., Pittsburgh PA, 1995.
29. Leban, I.; Golic, L. Acta. Pharm. Jugosl. 1988, 38, 71. 30. Bernardinelli, G.; Jefford, C. W.; Maric, D.; Thomson, C.;
Weber, J. Int. J. Quant. Chem. Bio. Sym. 1994, 21, 117.
TRAVAUX PRATIQUES D’ELECTRONIQUE 1ère année Semestre 1 Module GE11 et EN1 PRESENTATION DES COMPTES RENDUS DE TRAVAUX PRATIQUES. 2 TP2 : MONTAGES A AMPLIFICATEURS OPERATIONNELS . 5 EVALUATION DE TP A LA FIN DES 5 SEANCES DE TRAVAUX PRATIQUES PRESENTATION DES COMPTES RENDUS DE TRAVAUX PRATIQUES Chaque groupe rendra un rapport de TP sur copie double . L’entête des co
Daily Use of Whitening Strips on Tetracycline- Stained Teeth: Comparative CE 4 Results After 2 Months Gerard Kugel, DMD, MS Professor Dean for Research Abstract: This article reviews the efficacy of a new 6.5% hydrogen peroxide tooth-whitening gel strip for bleaching teeth that have been intrinsically stained Ayman Aboushala, DDS, MS from tetracycline. Given the severity of
 J Mol Model (2001) 7:26–33DOI 10.1007/s008940100013
Somsak Tonmunphean · Vudhichai Parasuk
J Mol Model (2001) 7:26–33DOI 10.1007/s008940100013
Somsak Tonmunphean · Vudhichai Parasuk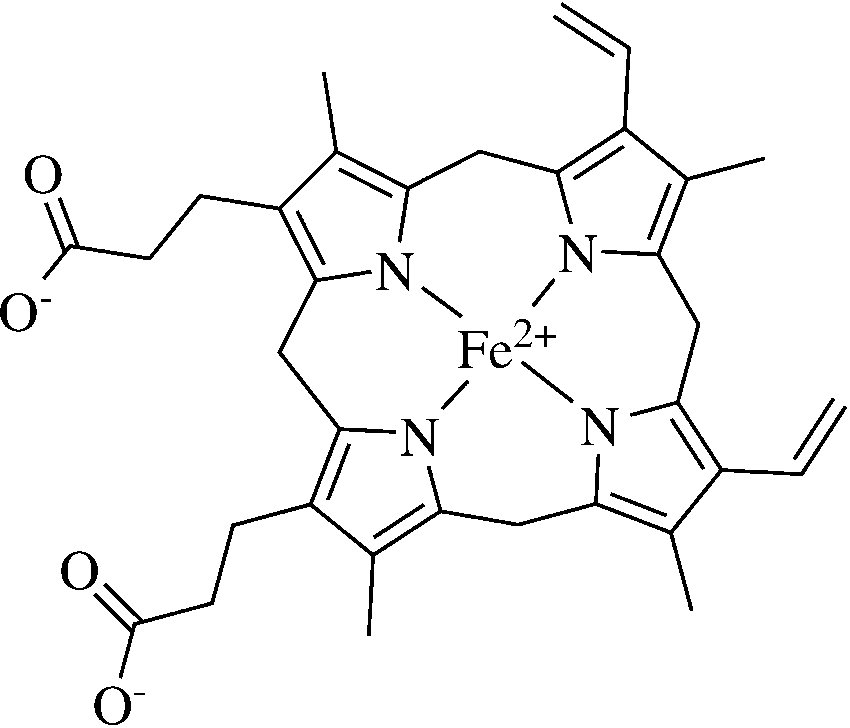
 O2 position [16] (Fig. 3). In pathway A, heme iron at-
results obtained will give information on how the chemi-
tacks the compound at the O2 position and produces a
cal structure of the drug should be modified to achieve
free radical at the O1 position. Later it rearranges to
suitable interactions. Hence, this could bring about the de-
form the C4 free radical. In pathway B, heme iron at-
velopment of new and more effective drugs.
O2 position [16] (Fig. 3). In pathway A, heme iron at-
results obtained will give information on how the chemi-
tacks the compound at the O2 position and produces a
cal structure of the drug should be modified to achieve
free radical at the O1 position. Later it rearranges to
suitable interactions. Hence, this could bring about the de-
form the C4 free radical. In pathway B, heme iron at-
velopment of new and more effective drugs.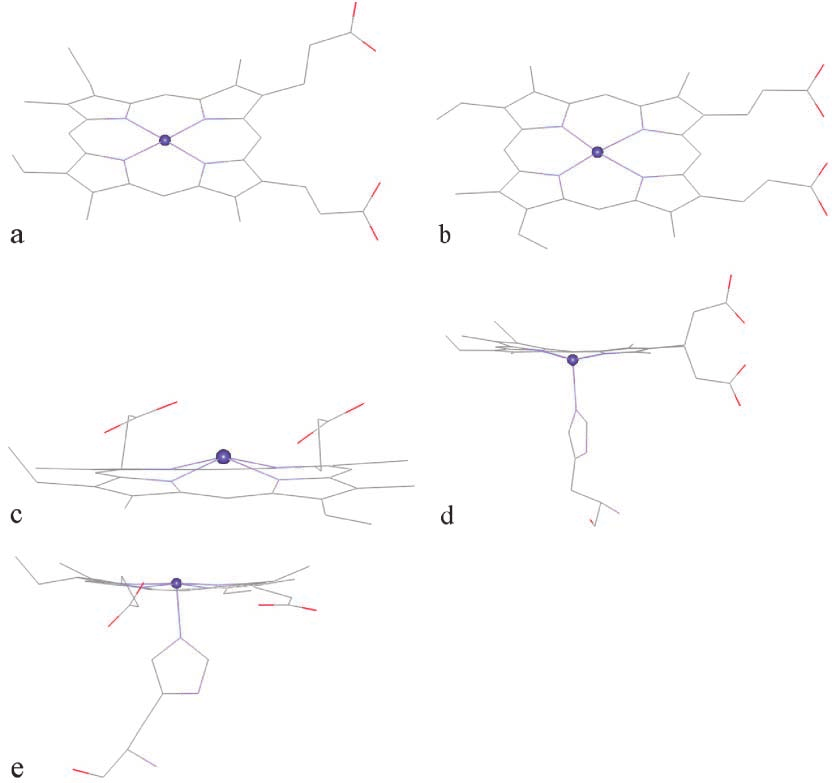 In docking calculations, the electrostatic potential is builtfrom atomic charges. Therefore, the choices for atomiccharges of both the ligand and receptor would have an effect on the docking results. Using charges obtained from ZINDO/S, HF/STO-3G, HF/3-21G, andHF/6-311G** levels of theory for heme-pdb, the dockingto the artemisinin X-ray structure with HF/3-21G
Fig. 4 The structures of the five heme compounds: (a) heme-pdb,
In docking calculations, the electrostatic potential is builtfrom atomic charges. Therefore, the choices for atomiccharges of both the ligand and receptor would have an effect on the docking results. Using charges obtained from ZINDO/S, HF/STO-3G, HF/3-21G, andHF/6-311G** levels of theory for heme-pdb, the dockingto the artemisinin X-ray structure with HF/3-21G
Fig. 4 The structures of the five heme compounds: (a) heme-pdb,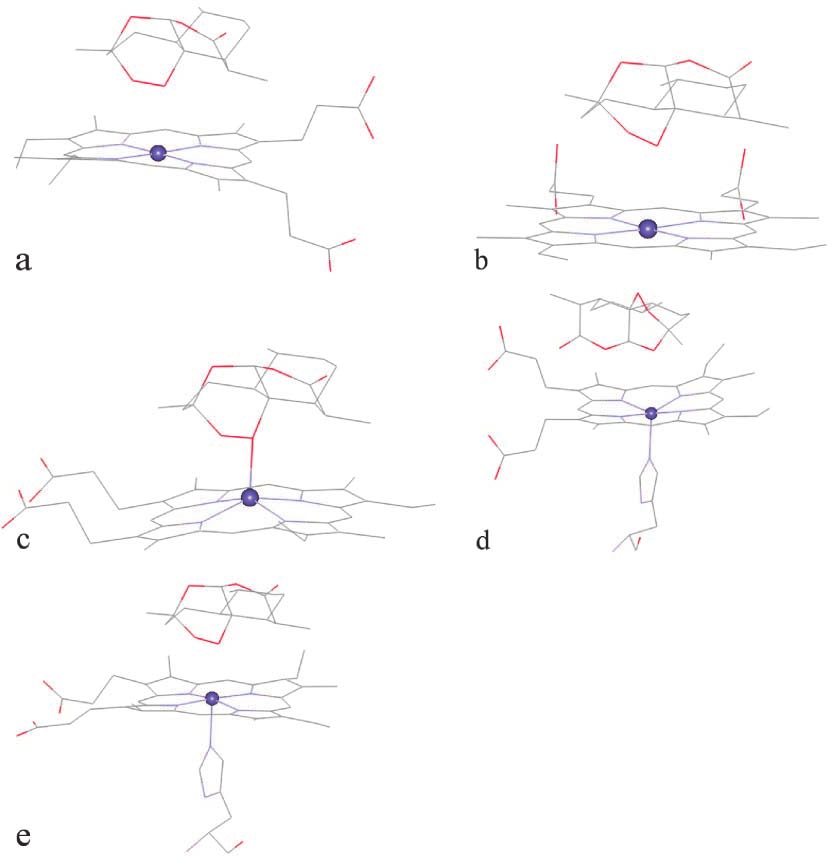 most occurring configuration, which has the shorterO13–Fe distance of 3.26 Å, compared with 5.95 and5.53 Å for O1–Fe and O2–Fe (Fig. 5d). Interestingly, thesecond most occurring configuration has shorter O1–Feand O2–Fe distances. Still, this distance is longer thanthose obtained from the docking with other heme struc-tures. For heme-oxy, the most occurring configurationhas O1–Fe as the shortest heme–artemisinin distancewith the binding energy of –32.32 kcal mol–1 (Fig. 5e).
most occurring configuration, which has the shorterO13–Fe distance of 3.26 Å, compared with 5.95 and5.53 Å for O1–Fe and O2–Fe (Fig. 5d). Interestingly, thesecond most occurring configuration has shorter O1–Feand O2–Fe distances. Still, this distance is longer thanthose obtained from the docking with other heme struc-tures. For heme-oxy, the most occurring configurationhas O1–Fe as the shortest heme–artemisinin distancewith the binding energy of –32.32 kcal mol–1 (Fig. 5e).